Nonlinear methods for linking climate to viral spread
Abstract
The effective population size, Nₑ(t), is a fundamental quantity in population genetics and phylodynamics: it captures genetic diversity and reveals demographic history through time. Coalescent‑based methods can reconstruct Nₑ(t) trajectories from time‑scaled phylogenies built from molecular sequence data, and linking those trajectories to external information — climate, epidemiological covariates, vector abundance — is essential for understanding the ecological and environmental drivers of pathogen population dynamics.
Existing coalescent frameworks that incorporate covariates almost always assume a log‑linear relationship between covariates and Nₑ(t). When the true relationship is nonlinear — as is biologically common — this assumption introduces bias and obscures real signal. We present a flexible Bayesian framework that integrates covariates into coalescent models with piecewise‑constant Nₑ(t) through a Gaussian process prior, naturally accommodating nonlinear effects without restrictive parametric assumptions and yielding interpretable uncertainty across the covariate space.
To balance global covariate‑driven patterns with local temporal dynamics, the Gaussian process prior is coupled with a Gaussian Markov random field that enforces smoothness in the Nₑ(t) trajectory itself. Efficient inference is achieved via Hamiltonian Monte Carlo over the high‑dimensional latent field, making the method computationally tractable for realistic genomic datasets.
We demonstrate the approach in simulation and on three empirical applications: yellow fever virus dynamics in Brazil (2016–2018), late‑Quaternary musk ox demography, and HIV‑1 CRF02 AG evolution in Cameroon. The framework confirms log‑linear relationships where appropriate but, crucially, reveals nonlinear covariate effects that previous methods miss or mischaracterise — including a sigmoidal relationship between temperature and YFV effective population size that goes undetected under standard log‑linear assumptions.
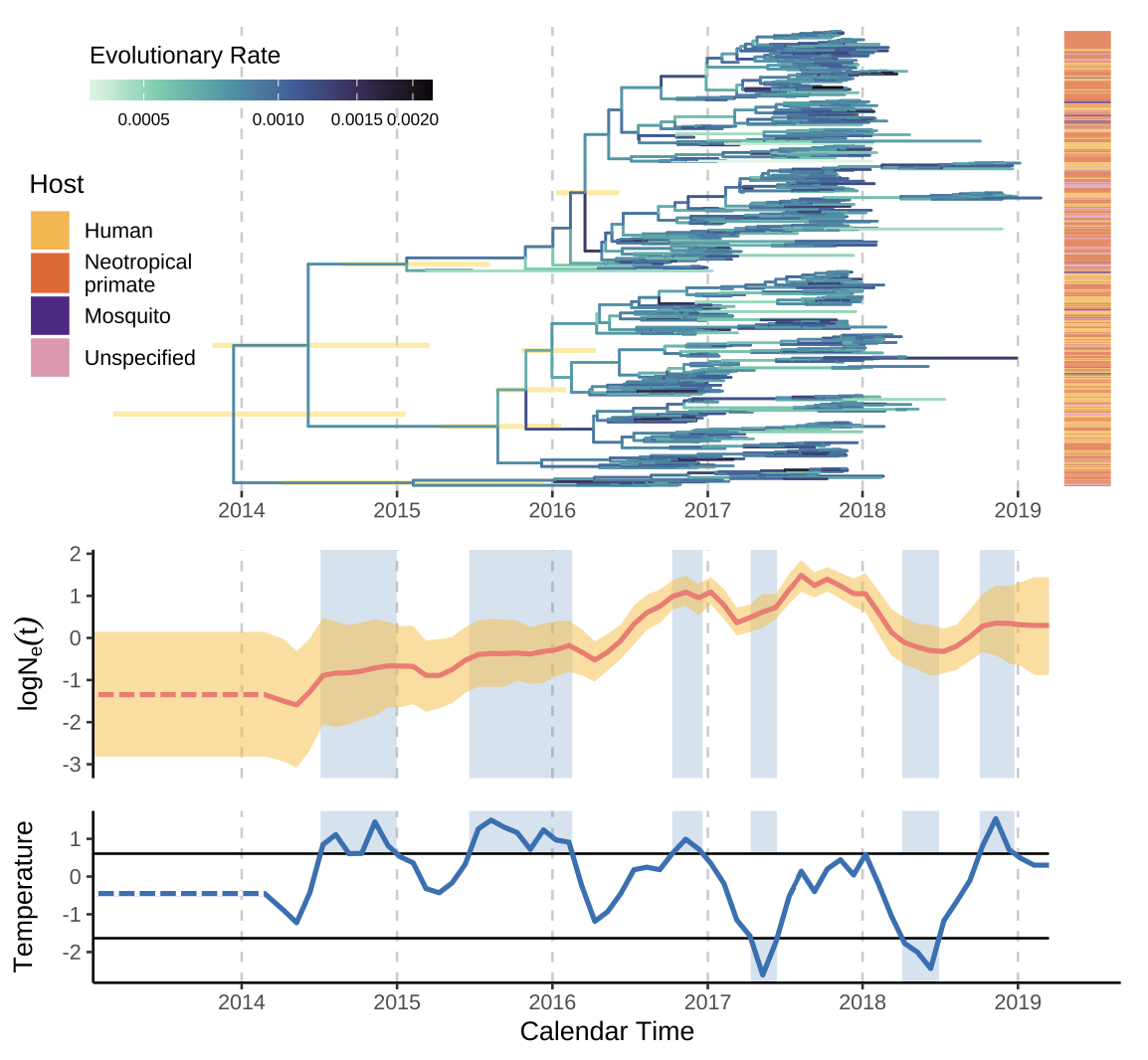
Figure. Yellow fever virus phylogeny and reconstructed effective population size in Brazil, 2016–2018. The upper panel shows the time‑scaled tree of YFV genomes from humans, non‑human primates and mosquitoes; the lower panel shows the inferred log Nₑ(t) trajectory together with average local temperature, illustrating the nonlinear relationship that the Gaussian‑process framework recovers but log‑linear models miss.
Context
Our new preprint, posted on arXiv, develops a more flexible Bayesian framework for one of the central problems in genomic surveillance: linking the rise and fall of a pathogen's effective population size to the environmental and epidemiological factors that drive it. Existing methods almost universally assume those drivers act log‑linearly on Nₑ(t) — a convenient but biologically unrealistic restriction. By replacing that assumption with a Gaussian‑process prior over the covariate space, the method recovers nonlinear, threshold‑like and saturating relationships that standard tools either flatten out or misattribute.
Applied to yellow fever virus in Brazil between 2016 and 2018, the framework uncovers a sigmoidal relationship between local temperature and YFV effective population size — a pattern fully consistent with vector and pathogen biology, but invisible under log‑linear modelling. For DeZi this matters directly: many of the climate‑sensitive questions about dengue, Zika, yellow fever and Oropouche transmission do not have linear answers, and methods that admit threshold and saturation effects are essential if genomic surveillance is to translate into useful predictions about how arboviruses respond to a changing climate.